

翻译:疯狂的技术宅
在本文中,我们将探讨如何通过用已编译的 WebAssembly 替换 JavaScript 来加速 Web 应用。
如果你还有听说过 WebAssembly,就先看一下解释:WebAssembly 是一种在浏览器中与 JavaScript 一起运行的新语言。没错, JavaScript 不再是唯一在浏览器中运行的语言了!
除了“不是 JavaScript”之外,最大的区别是你可以将 C/C++/Rust(甚至更多!)等语言的代码编译为 WebAssembly 并在浏览器中运行。因为 WebAssembly 是静态类型的,使用线性内存并以紧凑的二进制格式存储,所以它非常快,最终可以让我们以“接近原生”的速度运行代码,即速度接近你通过运行二进制文件达到的速度。能够利用现有工具和库在浏览器中使用的能力以及在运行速度上的潜力,是 WebAssembly 引人注目的两个原因。
到目前为止,WebAssembly 已被用于各种应用,从游戏(例如Doom 3)到把桌面程序移植到 Web(例如Autocad和 Figma )。它甚至可以在浏览器之外使用,例如 serverless 高效计算。
本文是一篇用 WebAssembly 对 Web 数据分析工具进行加速的研究性案例。为此我们用 C 编写的已有工具执行相同的计算,并将其编译为 WebAssembly 来替换慢速的 JavaScript 计算。
注意:本文深入研究了一些高级主题,比如编译 C 代码,但如果你没有相关经验,请不要担心;你仍然可以继续了解使用 WebAssembly 的可行性。
背景
我们将要使用的网络应用程序是fastq.bio,这是一个交互式的网络工具,可以让科学家快速预览 DNA 测序数据的质量;测序是读取 DNA 样品中“字母”(即核苷酸)的过程。
这是程序的截图:
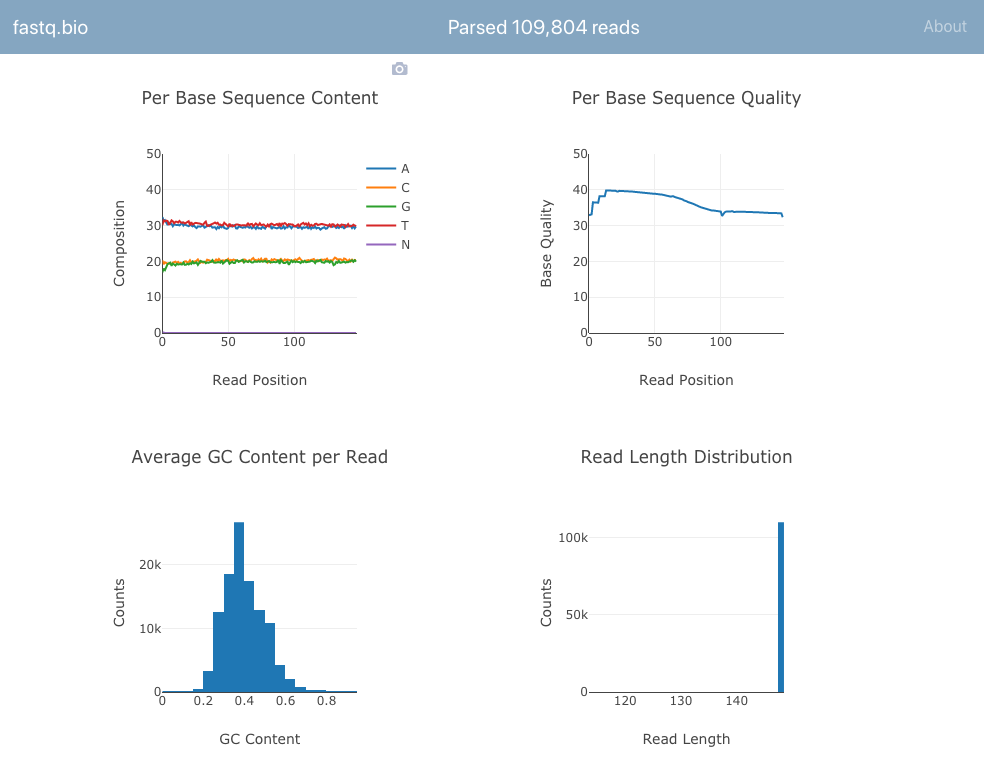
我们不会详细讨论关于计算的东西,但简而言之,上面的图表让科学家们了解了测序的进展情况,并能够一目了然地对数据的质量进行检查。
尽管许多命令行工具都能够生成这类质量控制报告,但 fastq.bio 的目标是在浏览器中提供数据质量的交互式预览。这对于不熟悉命令行的科学家特别有用。
该应用程序的输入是一个由测序仪器输出的纯文本文件,其中包含 DNA 序列列表和 DNA 序列中每个核苷酸的质量分数。由于该文件的格式称为“FASTQ”,因此网站的名称为 fastq.bio。
如果你对 FASTQ 格式感到好奇,请查看FASTQ的维基百科页面。 (警告:FASTQ文件格式可能会令你不忍直视。)
Fastq.Bio:JavaScript 实现
在 fastq.bio 的原始版本中,用户首先从计算机中选择 FASTQ 文件。使用 File 对象,程序先从随机位置读取一小块数据(使用FileReader API)。然后我们对这一大块数据,用 JavaScript 来执行基本的字符串操作并计算相关指标。这样的度量标准可以帮助我们跟踪在 DNA 片段的每个位置看到的 A,C,G 和 T 的数量。
一旦该数据块的度量标准计算完毕,我们将用 Plotly.js 以交互方式绘制结果,然后再转到文件中的下一个块。以小块处理文件的原因只是为了改善用户体验:一次处理整个文件需要太长时间,因为 FASTQ 文件通常有几百 GB。我们发现 0.5 MB 到 1 MB 之间的块大小会使程序运行得更加流程,并且可以更及时地向用户返回信息,但是这个数字会根据程序的具体情况和计算量的大小有所不同。
我们最开始用 JavaScript 实现的架构非常简单:
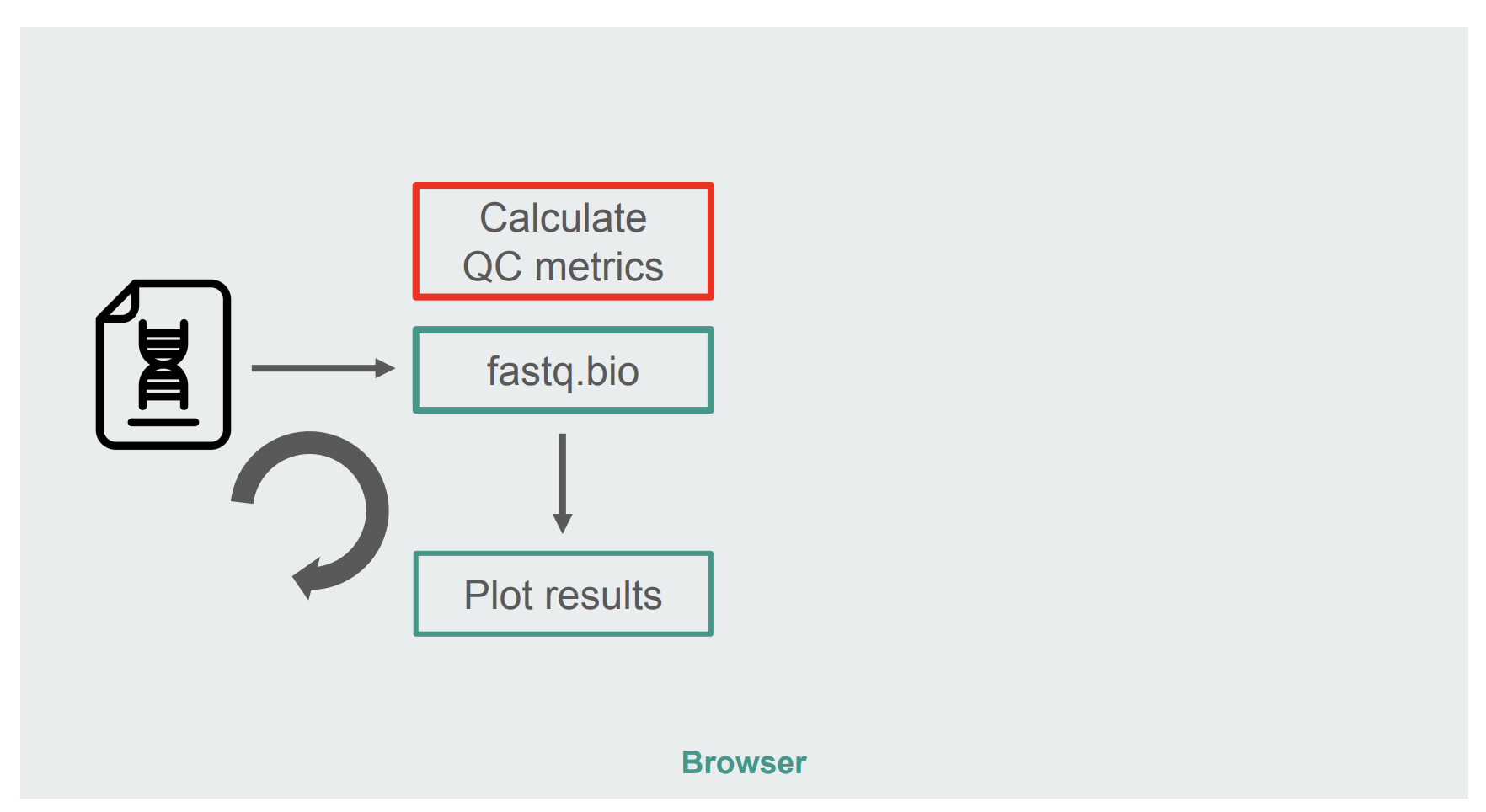
红色方框是进行字符串操作以生成指标的地方。该框是程序中计算密集度最高的那一部分,很显然应该用 WebAssembly 对其进行运行时优化。
Fastq.Bio:WebAssembly 实现
为了探索是否可以利用 WebAssembly 来加速 Web 应用,我们搜索了一个现成的工具来计算 FASTQ 文件的 QC 指标。具体来说,我们需要找一个用C/C++/Rust 编写的并且已经被科学界验证和信任得工具,然后把它移植到 WebAssembly。
经过一些研究,我们决定采用 seqtk,这是一个用 C 语言编写的常用开源工具,可以帮我们评估测序数据的质量。
在将其编译到 WebAssembly 之前,先让我们研究一下怎样将 seqtk 正常编译为二进制文件以便在命令行上运行。通过研究 Makefile,找到了用 gcc 进行编译的命令:
# Compile to binary
$ gcc seqtk.c \
-o seqtk \
-O2 \
-lm \
-lz
另一方面,为了将 seqtk 编译为 WebAssembly,我们需要用到 Emscripten 工具链,它可以直接替换现有的构建工具,使编译 WebAssembly 的工作更容易。如果你没有安装 Emscripten,可以下载我们上传到 Dockerhub 上的 docker 镜像,该镜像中包含了你需要的工具(你也可以从头开始安装,但这样需要你花一点时间时间):
$ docker pull robertaboukhalil/emsdk:1.38.26
$ docker run -dt --name wasm-seqtk robertaboukhalil/emsdk:1.38.26
在容器内部,我们可以使用 emcc 编译器替代 gcc:
# Compile to WebAssembly
$ emcc seqtk.c \
-o seqtk.js \
-O2 \
-lm \
-s USE_ZLIB=1 \
-s FORCE_FILESYSTEM=1
如你所见,编译成二进制可执行文件和 WebAssembly 的方法之间的差异很小:
- 我们要用 Emscripten 生成一个
.wasm和一个.js来对 WebAssembly 模块进行实例化,而不是输出一个二进制可执行文件seqtk。 - 为了支持 zlib 库,我们用了
USE_ZLIB标志。zlib 库很常见,已经被移植到了 WebAssembly 中,Emscripten 会在我们的项目中包含它 - 我们启用 Emscripten 的虚拟文件系统,这是一个类似 POSIX 的文件系统(源代码),但是它只运行在浏览器的 RAM 中,并在刷新页面时消失(除非你用了 IndexedDB 在浏览器中保存其状态,但这不是本文所要研究的内容)。
为什么要启用虚拟文件系统?要回答这个问题,先让我们比较一下在命令行调用 seqtk 和用 JavaScript 调用已编译的 WebAssembly 模块这两种方式:
# 在命令行调用
$ ./seqtk fqchk data.fastq
# 在浏览器控制台中调用
> Module.callMain(["fqchk", "data.fastq"])
虚拟文件系统非常强大,因为这意味着不必为了处理输入参数而重写 seqtk 。我们可以将一块数据作为文件 data.fastq 挂载到虚拟文件系统上,然后简单地调用 seqtk 的 main()函数即可。
将 seqtk 编译为 WebAssembly 后,得到了新的 fastq.bio 架构:
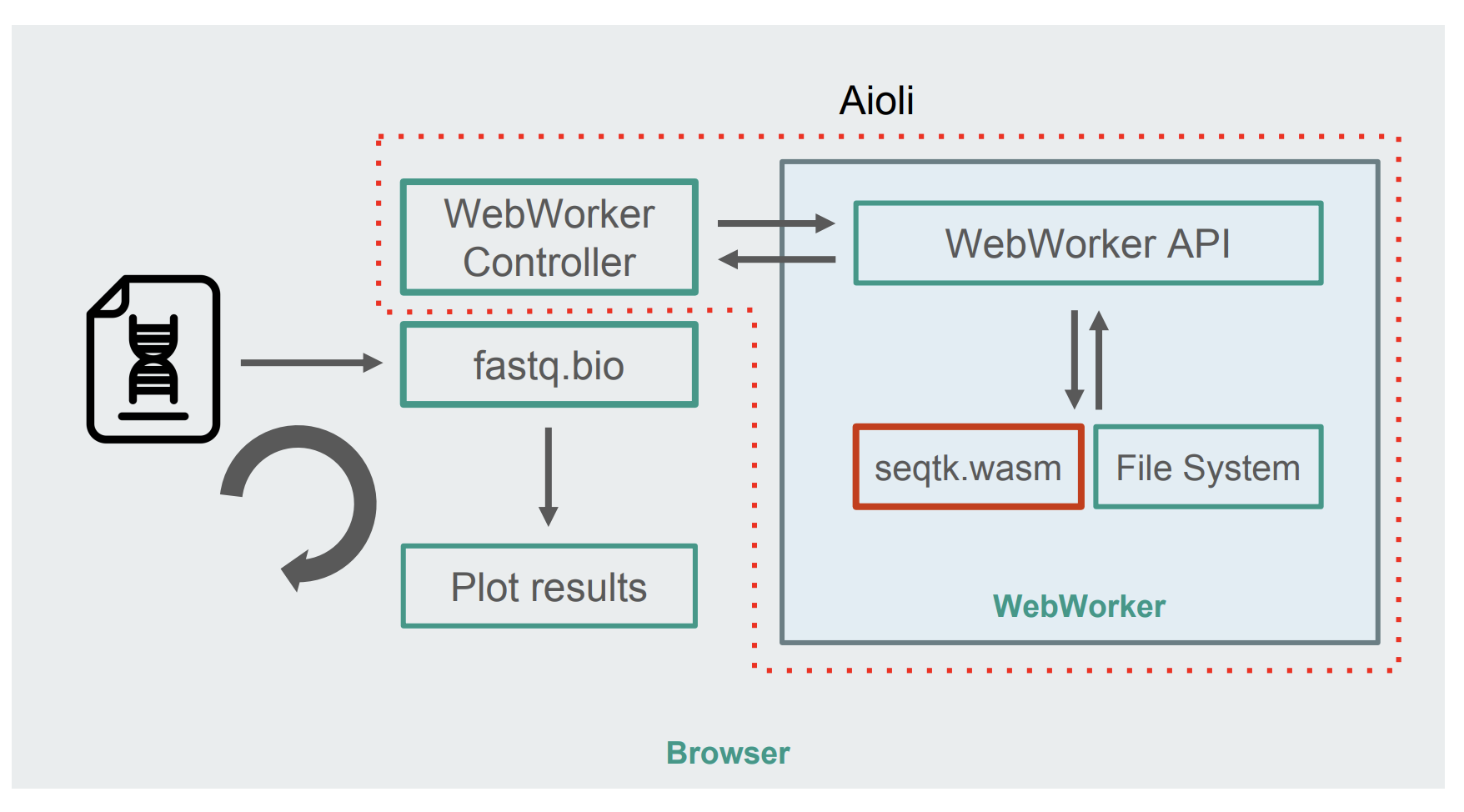
webAssembly 的体系结构和 fastW.bio 的 WebWorkers 实现:在输入文件中随机抽样,用 WebAssembly 在WebWorker 中计算指标,绘制结果并循环
如图所示,不用浏览器主线程而是用 WebWorkers ,这样可以在后台线程中执行我们的计算,并避免对浏览器的响应性产生负面影响。具体来说,WebWorker 控制器启动 Worker 并管理与主线程的通信。对于 Worker,API 执行它收到的请求。
然后我们可以要求 Worker 对刚挂载的文件运行 seqtk 命令。当 seqtk 完成运行时,Worker 通过 Promise 将结果发回主线程。收到消息后,主线程用结果输出来更新图表。与 JavaScript 版本类似,我们用块的形式去处理文件,并在每次循环时更新可视化图表。
性能优化
为了评估 WebAssembly 是否真的能够提高运行效率,我们用每秒读取并处理的数量作为度量指标来比较 JavaScript 和 WebAssembly 两种实现。在这里忽略了生成交互式图表所需的时间,因为两种实现都用了 JavaScript 来达到这一目的。
开箱即用,可以看到速度大约提升了 9 倍:
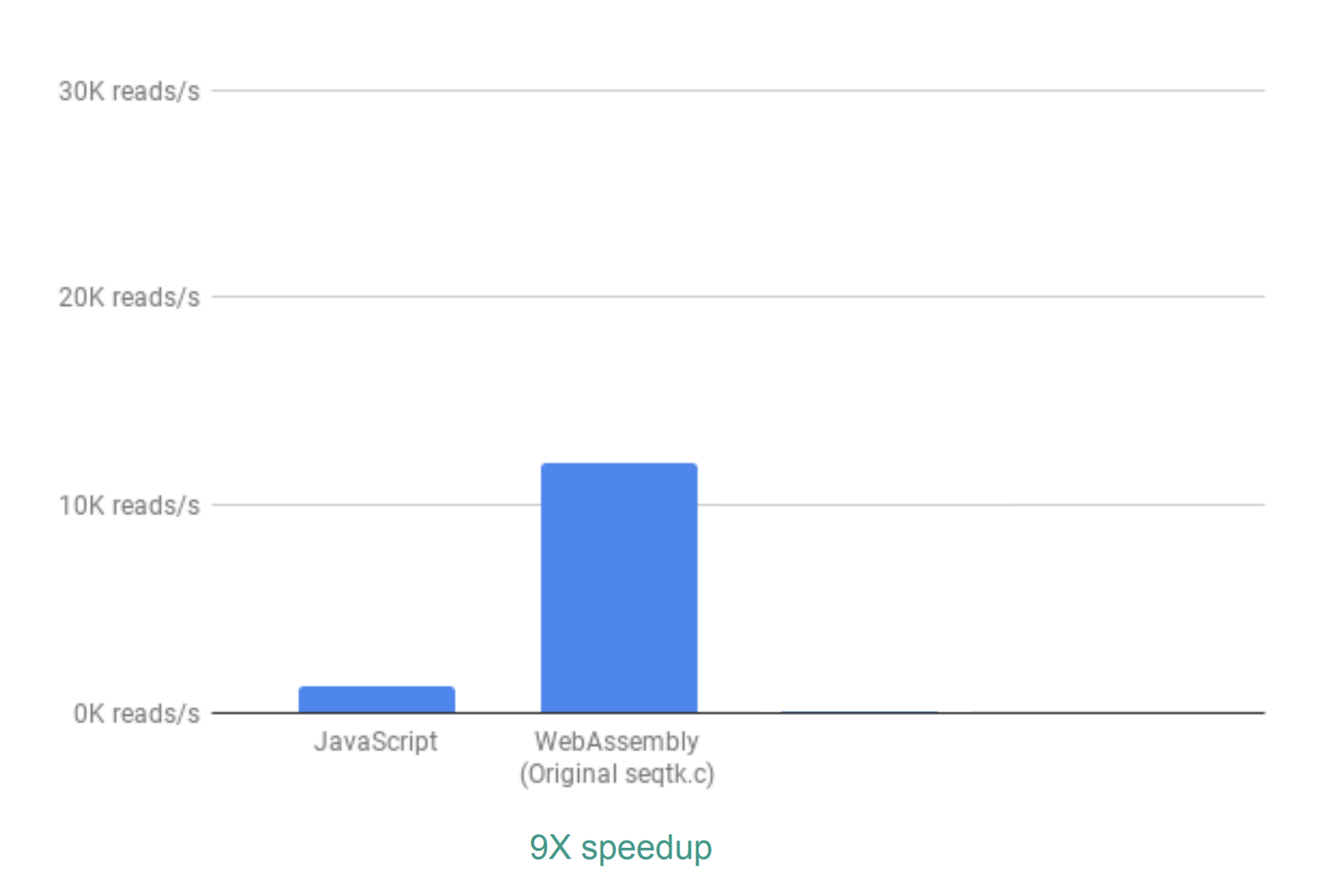
WebAssembly 与 JavaScript 实现相比,速度提升了 9 倍
这样已经很好了,因为它相对容易实现(前提是你理解了 WebAssembly!)。
接下来,我们注意到虽然 seqtk 输出了许多有用的QC指标,但程序实际上并未使用或绘制了这些指标。通过剔除不需要的指标输出,可以看到速度提高 13 倍:
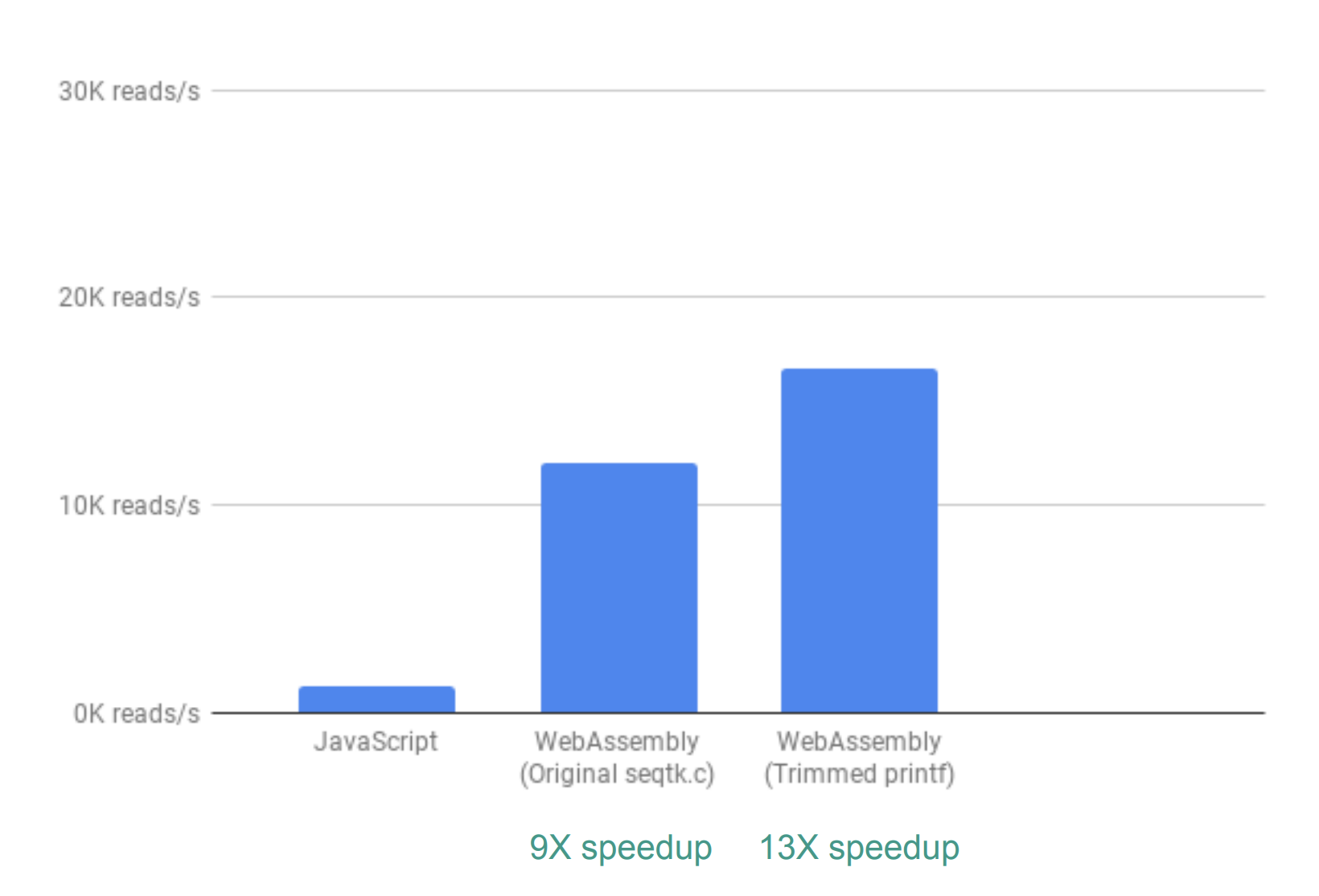
删除不必要的输出可以进一步提高性能。
实现它是多么的容易,这又是一个很大的改进。
最后,我们还会进一步改进。到目前为止,fastq.bio 通过调用两个不同的C函数来获取感兴趣的指标,每个函数计算一组不同的指标。具体做法是一个函数以直方图的形式返回信息(即被列入范围的值的列表),而另一个函数返回 DNA 序列位置的信息。不幸的是这意味着同一块文件被读取了两次,这是没有必要的。
所以我们把这两个函数的代码合并为一个(可以不用去修改 C 代码!)。由于两个输出的列数不同,我们在 JavaScript 这边做了一些重构。这是值得的:可以让我们得到 20 倍的速度提升!
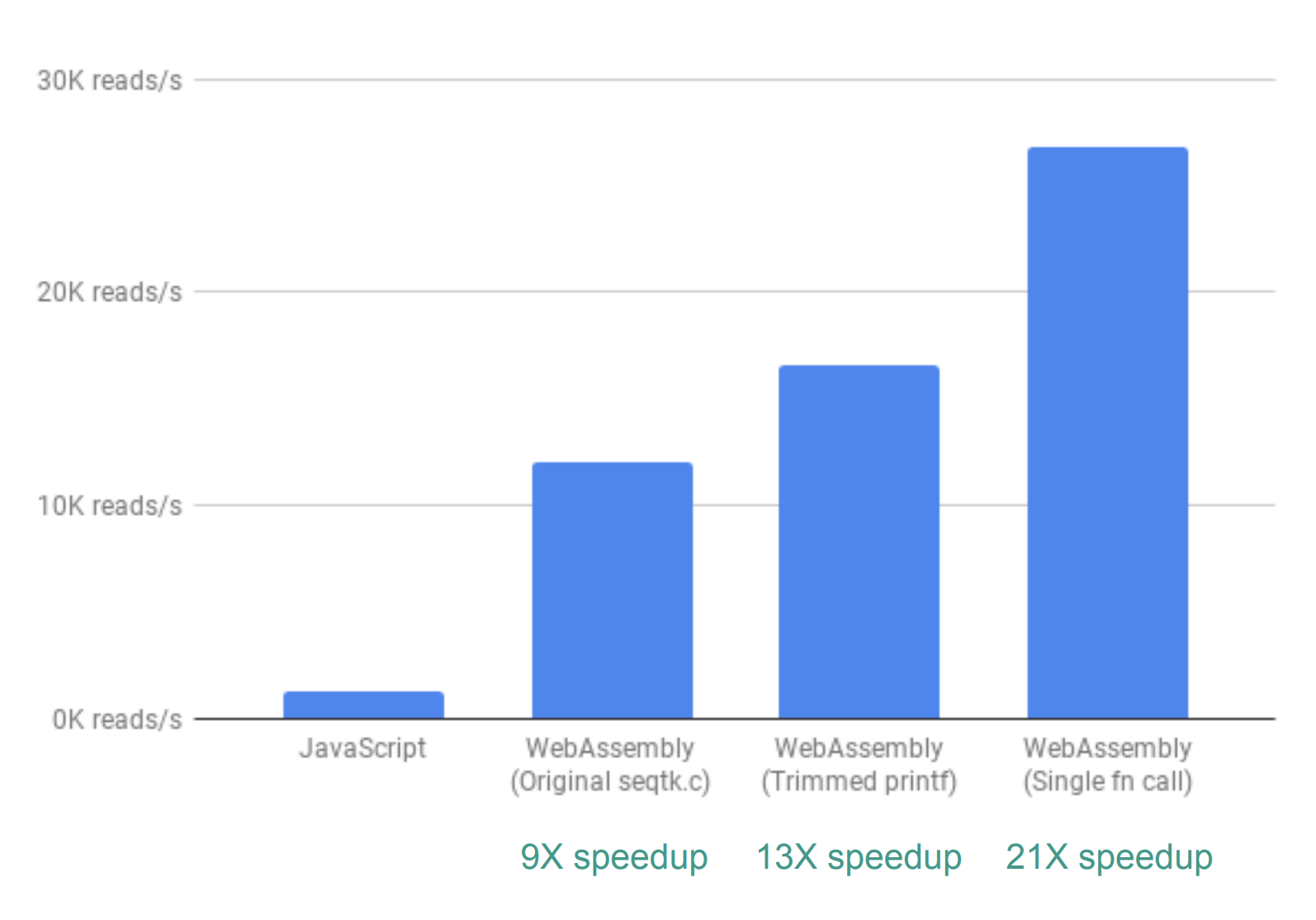
小心
使用 WebAssembly 时,不要期望总是获得 20 倍的加速。如果在内存中加载非常大的文件,或者需要在 WebAssembly 和 JavaScript 之间进行大量通信,则可能会变慢。你可能只会得到 2 倍甚至是 20% 的速度。
结论
我们已经看到,通过调用编译的 WebAssembly 来替换 JavaScript 可以使处理速度显著增加。由于这些计算所需的代码已经存在于 C 中,因此我们得到了重用可信工具带来的额外好处。正如前面所提到的,WebAssembly 并不总是适合这种工作,所以还需要明智地去使用它。
欢迎关注公众号:前端先锋,获取更多前端干货。

